Chemical Space Coverage
Abstract
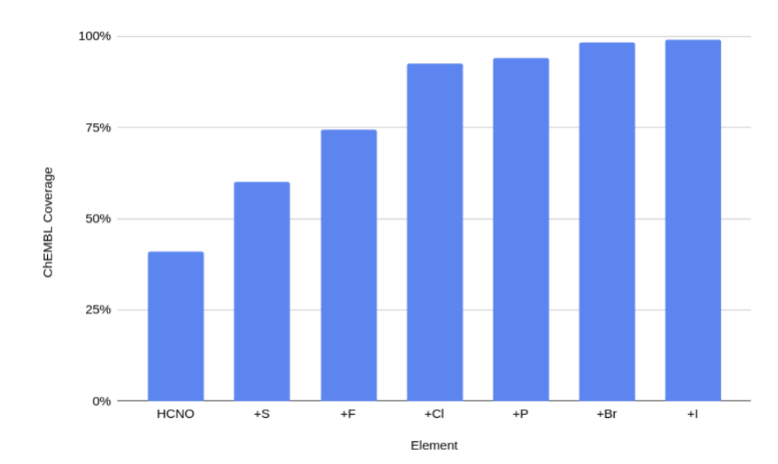
We have developed a neural network potential energy function for use in drug discovery, with chemical element support extended from 41% to 94% of druglike molecules based on ChEMBL. We expand on the work of Smith et al. with their highly accurate network for the elements H, C, N, O, creating a network for H, C, N, O, S, F, Cl, P. We focus particularly on the calculation of relative conformer energies, for which we show that our new potential energy function has an RMSE of 0.70 kcal/mol for prospective druglike molecule conformers, substantially better than the previous state of the art. The speed and accuracy of this model could greatly accelerate the parameterization of protein-ligand binding free energy calculations for novel druglike molecules.
Table 1
RMSEs (kcal/mol) of ANI-1x and Schr ̈odinger-ANI, each judged relative to its own reference level of theory (ωB97X/6-31G* and ωB97X-D/6-31G* respectively). Genentech RMSEs are for single-point energies, while missing torsion RMSEs are for geometries optimized with the respective model.
| Test Set | ANI-1x | Schrodinger-ANI |
|---|---|---|
| Genentech (HCNO) | 0.50 | 0.37 |
| Missing Torsions (HCNO) | 1.07 | 0.69 |
| Genentech (all) | N/A | 0.38 |
| Missing Torsions (all) | N/A | 0.70 |